

The European Parliament (EP) has published two draft reports on the Commission’s proposals for a new Pharmaceutical Regulation and Directive.
The reports have been drafted by the MEPs assigned as rapporteurs for the Directive (Pernille Weiss, EPP) and for the Regulation (Tiemo Wölken, S&D) and will be debated at the Committee for the Environment, Public Health and Food Safety (ENVI) later this month or beginning of November.
The report on the Directive is favourable to the innovative pharmaceutical industry and proposes amendments aimed at creating “an attractive environment for research, development and production of medicines in the Union”, whilst the report on the Regulation tightens the obligations on the industry and focuses on ensuring “greater accessibility, affordability, and availability of medicinal products for patients across the whole Union.”
Below we give an overview of the main amendments being proposed in both draft reports.
EP Draft report on the Directive: Focus on Promoting R&D and Innovation
The report on the Directive contains amendments clearly aimed at promoting research and innovation, and includes proposals that represent a significant turnaround from the Commission’s positioning on sensitive topics such as regulatory data protection, access to medicines and protection of the environment.
- Regulatory data protection
One of the most significant elements of the Commission’s Proposal is the reduction of the standard period of regulatory data protection from the (current) 8 years to 6 years meaning that companies would be entitled to 8 (6+2) years of protection instead of the current 10.
By contrast, the EP’s draft report proposes an extension of the regulatory data protection period to 9 years so that companies would be entitled to 11 (9+2) yearsof protection in total.
In addition, the report deletes the requirement to launch in all Member States (MS) for companies wishing to obtain 2 additional years of data protection. It also prolongs some of the extensions of data protection foreseen in the Commission’s Proposal.
The table below summarises these changes:
| EC Proposal | EP Draft | |
| Standard period of protection (data + market protection) | 6+2 years | 9+2 years |
| Product released and supplied in sufficient quantities in MS where the marketing authorisation (MA) is valid | + 2 years data protection | Deleted (see below under ‘access to medicines’ for new provision introduced instead) |
| Product addresses unmet medical need | + 6 months data protection | + 1 year data protection |
| Comparative clinical trials conducted in accordance with EMA scientific advice | + 6 months data protection (only for initial applications containing a new active substance (NAS)) | + 6 months (for all applications and product doesn’t need to contain a NAS) |
| New therapeutic indication showing significant clinical benefit | + 1 year data protection (prolongation can only be granted once) | + 1 year data protection (prolongation can only be granted once) |
| Other periods of data protection: | ||
| Repurposed medicinal products | 4 years data protection | 4 years data protection |
Notably, the EP report also cuts down on other measures that the Commission had introduced to promote generic access to the market such as the expansion of the bolar exemption. The EP report proposes to limit the bolar exemption to approval submissions only (as already foreseen in the current legislation), and not to cover additional activities such as HTA assessment or pricing and reimbursement as proposed by the Commission.
- Access to medicines
The EP’s report separates the promotion of access to medicines from regulatory data protection periods.
Instead of the + 2 year RDP extension foreseen in the Commission Proposal when a medicine is launched in all relevant Member States, the EP’s draft intends to strengthen access to medicines via a new provision.
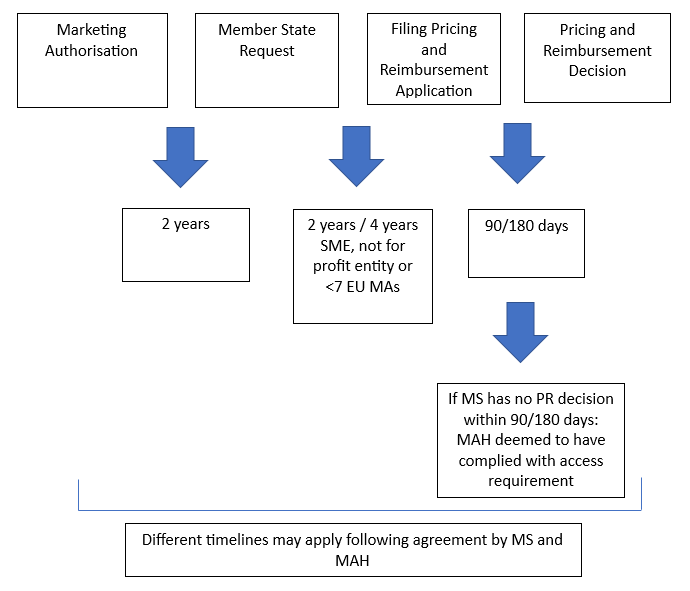
According to this provision, companies will be required to apply for pricing and reimbursement in all those Member States that request that the product is launched in their territory. Member States must raise such request within 2 years following the marketing authorisation. Companies will have a period of 2 years to comply with the requests or 4 years if the company concerned is an SME, a not-for-profit entity or has fewer than 7 centrally authorised products. Failure to comply with this requirement shall be subject to the imposition of financial penalties.
The graph below shows the overall access timeline, including the timeline of 90 or 180 days for obtaining a pricing and reimbursement decision from the Member States imposed in accordance with the Transparency Directive[1].
- Paediatric rewards
The report also foresees a new+ 1 year extension of the supplementary protection certificate where the paediatric investigation plan is conducted for a disease that is different from the one for which the medicinal product is intended in the adult population.
- Unmet Medical Need (UMN)
Another measure in the EP’s report is the broader UMN definition to the extent that it covers progressive diseases and medicines that can have a meaningful positive impact on quality of life or delay the onset of the disease or its complications. The Commission’s proposal focuses on high morbidity or mortality only and this is seen as too restrictive by many.
- Environmental risk assessment
Departing from the Commission’s proposal, the European Parliament’s report proposes that a negative environmental risk should not automatically lead to the refusal of a marketing authorisation.
Draft report on the Regulation: Focus on Greater Access, Affordability and Availability
In stark contrast with the Directive’s report, the main drivers for the EP’s report on the Regulation are ensuring greater accessibility, affordability, and availability. With these objectives in mind, the report reduces orphan market exclusivity periods and maintains the obligations the Commission proposes to impose on companies to ensure continuity of supply and prevention of shortages.
In addition to that, the EP’s report proposes to get rid of the transferable exclusivity vouchers the Commission had introduced for priority antimicrobials. It also removes the regulatory sandboxes which were aimed at supporting the development of highly innovative medicines.
- Orphan incentives
The Commission’s proposal reduced the standard period of orphan market exclusivity from 10 to 9 years. The EP’s report proposes to reduce this period even further, bringing it down to 8 years.
Other periods of orphan market exclusivity foreseen in the Commission’s proposal are maintained with some slight amendments shown in the table below.
| EC Proposal | EP Draft | |
| Standard orphan market exclusivity | 9 years | 8 years |
| Orphan products addressing a high unmet medical need (HUMN) | 10 years | 10 years (HUMN defined more tightly) |
| New orphan therapeutic indication of products already authorised | Not specified | 5 years |
| Well established use orphan medicinal products | 5 years | 3 years |
| Extensions of orphan market exclusivity: | ||
| Orphan medicine released and supplied in sufficient quantities in relevant MS | + 1 year | Deleted |
| New indication for a different orphan condition | + 1 year (if indication obtained at least 2 years before the end of exclusivity period) | +1 year (if indication obtained at least 3 years before the end of exclusivity period) |
In addition, the report reintroduces the possibility to reduce the standard periods of orphan market exclusivity foreseen above to 5 years if it is established that the orphan designation criteria are no longer met. The Commission had proposed to abolish this measure from the current legislation, and the EP report proposes to reintroduce it.
Most significantly, the EP’s report does not contest the weaker form of protection proposed by the Commission whereby orphan market exclusivity shall not prevent the submission and assessment of a marketing authorisation application for a similar medicinal product, including generics or biosimilars, where the remainder of the duration of the market exclusivity is less than 2 years.
See here for greater detail on this measure and the Commission’s proposals for orphan medicines.
- HUMN definition
The HUMN definition is tightened so it applies to orphan products that result in a “substantial” reduction of disease morbidity or mortality as opposed to the “meaningful” reduction that the Commission had proposed. As such, this amendment travels in the opposite direction of the amended definition of UMN under the Directive.
- Transferable exclusivity vouchers for antimicrobials
The EP’s report abolishes the transferable exclusivity vouchers foreseen in the Commission’s proposal for priority antimicrobials. Instead, the report proposes the establishment of the ‘European Medicines Facility’ as a Union Agency that conducts research and development on antimicrobials and other areas of unmet need.
- Regulatory sandbox
The report also proposes to remove Commission proposals aimed at supporting faster access to innovative technologies such as the regulatory sandbox. The rapporteur claims the regulatory sandbox would introduce a parallel regulatory framework that could pave the way for the circumvention of rules and obligations laid down in other frameworks.
Next steps
These reports represent the views of the rapporteurs leading the discussions on the Directive and the Regulation. The reports will be subject to debate at the ENVI Committee (first meeting expected soon in October/November). After one or more meetings, the Committee should eventually adopt a consolidated report and this report would then be subject to the EP’s plenary vote. Given the diametrically opposed views reflected in these two reports, and the sensitivity and breadth of the topics covered in the package we expect a heated debate.
The Council will examine the Commission’s proposals in parallel to the EP. We are also expecting to see conflicting views within the Council with some Member States (e.g. Germany or Denmark) favouring the promotion of innovation and others (e.g. Estonia or Malta) defending access and affordability and availability of medicines.
Overall, the sensitivity of the measures contained in the proposals and the divergence in views that we are already seeing around them show that this will be a very complex legislative process and the Parliamentary elections in June 2024 will only bring added pressure to the timings and negotiations.
We continue to closely follow the detail of the Package. Further insight can be viewed here .
[1] Article 2 of the Directive provides that Member States shall ensure that a decision on the price which may be charged for a medicinal product is adopted within 90 days of the receipt of an application. In accordance with Article 6(1) of the Directive, the same timeline applies with respect to the decision on reimbursement. Article 6(2) of the Directive provides that where a Member State does not permit an application to be made for reimbursement before the competent authorities have agreed the price to be charged for the product, the Member State concerned shall ensure that the overall period of time taken by the two procedures does not exceed 180 days. Clock-stops may extend the timelines.






